
Le projet PERIGENOMED

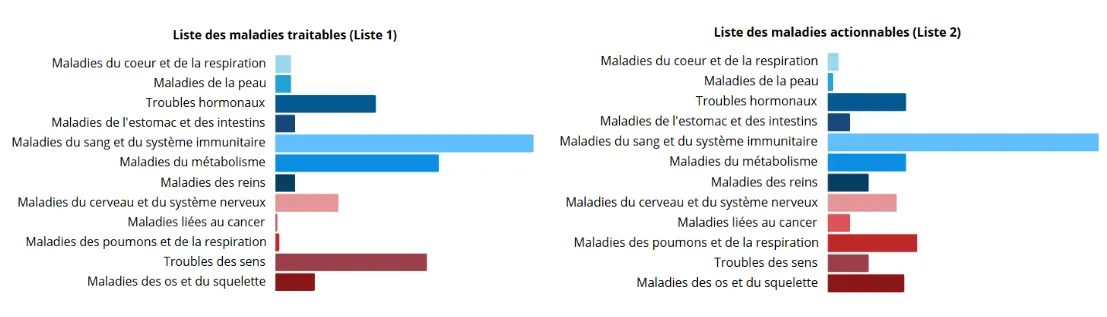
Maladies dépistées dans PERIGENOMED-CLINICS 1 (PGC1)
La première phase du projet PERIGENOMED, PERIGENOMED-CLINICS 1 (PGC1), prévoit d'étudier 2 listes de maladies différentes.
La liste principale du projet comporte 172 groupes de maladies traitables. Ce sont des maladies pour lesquelles il existe un traitement ou une action préventive, par exemple un régime alimentaire adapté, permettant d'éviter ou de limiter fortement les conséquences de la maladie chez la majorité des enfants.
La deuxième liste, optionnelle, comporte 218 groupes de maladies actionnables. Pour ces maladies, une action médicale préventive peut permettre de modifier l'évolution de la maladie et d'améliorer la santé chez certains enfants. Il se peut aussi que des traitements soient en cours de développement ou fassent l'objet d'études cliniques.
Version de la liste en date du 11/06/2025.
Maladies du coeur et de la circulation sanguine (cardiovasculaire)
- Syndrome de Jervell et Lange-Nielsen (KCNE1, KCNQ1)
- Syndrome du QT long (CALM1, CALM2, CALM3, KCNH2, KCNQ1)
- Syndrome de Timothy (CACNA1C)
Maladies de la peau (dermatologie)
- Xeroderma pigmentosum (défaut de réparation de l'ADN) (DDB2, ERCC2, ERCC3, ERCC4, ERCC5, POLH, XPA, XPC)
Troubles hormonaux (endocrinologie)
- Abétalipoprotéinémie (MTTP)
- Déficit isolé en ACTH (TBX19)
- Hyperplasie congénitale des surrénales (CYP21A2, HSD3B2)
- Hypoplasie congénitale des surrénales (NROB1)
- Différence du développement sexuel 46,XY- insuffisance surrénalienne par déficit en CYP11A1 (CYP11A1)
- Adrénoleucodystrophie (ABCD1)
- Syndrome de Bamforth-Lazarus (FOXE1)
- Maladie de rétention des chylomicrons (SAR1B)
- Diabète insipide (AVP)
- Diabète néonatal (ABCC8, GLIS3, KCNJ11)
- Déficit familial en apolipoprotéine C-II (APOC2)
- Déficit isolé familial en glucocorticoïdes (MC2R, MRAP, NNT)
- Hypercholestérolémie familiale (LDLR, LDLRAP1, PCSK9)
- Hyperinsulinisme congénital (ABCC8, GCK, GLUD1, KCNJ11)
- Hyperlipoprotéinémie (GPIHBP1)
- Hyperparathyroïdie néonatale sévère (CASR)
- Hypocalcémie (CASR)
- Hypothyroïdie centrale avec macroorchidie (IGSF1)
- Hypothyroïdie congénitale sans goitre (IRS4, NKX2-5, PAX8, TBL1X, THRA, TRHR, TSHB, TSHR)
- Syndrome de chylomicronémie familiale (LMF1)
- Hyperplasie congénitale lipoïde des surrénales (STAR)
- Néoplasies endocriniennes multiples (RET)
- Adénome hypophysaire (GPR101)
- Obésité par déficit en proprotéine convertase 1/3 (PCSK1)
- Pseudopseudohypoparathyroïdisme (GNAS)
- Sitostérolémie (ABCG5, ABCG8)
- Dyshormonosynthèse thyroïdienne familiale (DUOX2, DUOXA2, IYD, SLC5A5, TG, TPO)
Maladies de l'estomac et des intestins (gastroentérologie)
- Déficit congénital de synthèse des acides biliaires (AKR1D1, HSD3B7)
- Cholestase intrahépatique familiale progressive (ABCB11, ABCB4, ATP8B1, MYO5B, TJP2)
- Syndrome de Crigler-Najjar (type I et II) (UGT1A1)
- Diarrhée chronique congénitale avec entéropathie exsudative (DGAT1)
- Hypertriglycéridémie (GPD1)
Maladies du sang et du système immunitaire (hématologie / immunologie)
- Agammaglobulinémie (BTK, PIK3R1)
- Thrombopénie congénitale amégacaryocytaire congénitale (MPL, THPO)
- Déficit en glucose-6-phosphate déshydrogénase classe I (G6PD)
- Anémie dysérythropoïétique liée à l'X avec plaquettes anormales et neutropénie (GATA1)
- Syndrome de Bernard-Soulier (GP1BA, GP1BB, GP9)
- Bêta-thalassémie (HBB)
- Macrothrombocytopénie autosomique dominante (ACTN1)
- Syndrome de Chédiak-Higashi (LYST)
- Granulomatose chronique (CYBB, NCF4)
- Déficits immunitaires combinés avec granulomes (RAG2)
- Immunodéficience combinée (MTHFD1, RAG1)
- Syndrome d'hyperactivation du complément-thrombose-entéropathie avec perte de protéines (CD55)
- Anémie de Diamond-Blackfan (GATA1, HEATR3, RPL11, RPL15, RPL18, RPL26, RPL27, RPL31, RPL35A, RPL5, RPS10, RPS15A, RPS17, RPS19, RPS24, RPS26, RPS27, RPS28, RPS29, RPS7, TSR2)
- Anémie de Fanconi (FANCA)
- Malabsorption héréditaire de l'acide folique (SLC46A1)
- Dysplasie hématodiaphysaire de Ghosal (TBXAS1)
- Thrombasthénie de Glanzmann (ITGA2B, ITGB3)
- Granulomatose chronique (CYBA, CYBC1, NCF1, NCF2)
- Lymphohistiocytose familiale (PRF1, STX11, STXBP2, UNC13D)
- Hémophilie (F8, F9)
- Syndrome hyper-IgE avec infections récurrentes (DOCK8)
- Déficit immunitaire combiné par déficit en RAG1/2 (FOXP3, RAG1, RAG2)
- Immunodéficience (ARPC1B, ARPC5, BCL11B, CARD9, CD28, CD3D, CD3G, CD40, CD40LG, CORO1A, DNMT3B, HELLS, IFNGR1, IKBKB, IL21R, IL2RA, IL7R, LCK, MAGT1, MALT1, NHEJ1, ORAI1, PIK3CD, PIK3R1, POLD3, PRKDC, PTPRC, SASH3, SLC19A1, STIM1, STK4, ZAP70, ZBTB24)
- Déficit immunitaire par déficit d'expression des molécules CMH de classe I (B2M)
- Déficit immunitaire combiné par déficit en CARD11 (CARD11)
- Déficit immunitaire combiné sévère T-B+ par déficit en CD3delta/epsilon/zêta (CD3E)
- Granulomatose chronique (CYBB)
- Déficit d'adhésion leucocytaire (ITGB2)
- Syndrome LIG4 (LIG4)
- Syndrome lymphoprolifératif (SH2D1A)
- Déficit du complexe majeur d’histocompatibilité (CIITA, RFX5, RFXANK, RFXAP)
- Neutropénie sévère congénitale (G6PC3, HAX1)
- Syndrome d’Omenn (DCLRE1C, RAG1, RAG2)
- Anémie hémolytique par déficit en pyruvate kinase du globule rouge (PKLR)
- Syndrome de synostose radio-ulnaire-thrombocytopénie amégacaryocytique (MECOM)
- Déficit rare en facteurs de coagulation (F10, F13A1, F13B, F7)
- Dysgénésie réticulaire (AK2)
- Déficit immunitaire combiné sévère (SCID) (ADA, DCLRE1C, IL2RG, JAK3, RAG1, RAG2)
- Drépanocytose (HBB)
- Déficit en lymphocytes T et lymphopénie (FOXN1)
- Thrombopénie (GATA1, MPIG6B)
- Purpura thrombotique thrombocytopénique congénital (ADAMTS13)
- Déficit en transcobalamine II (TCN2)
- Maladie de von Willebrand (VWF)
- Syndrome de Wiskott-Aldrich (WAS, WIPF1)
Maladies du métabolisme
- Acidurie 3-hydroxy-3-méthylglutarique (HMGCL)
- Déficit en 3-hydroxy-3-méthylglutaryl-CoA synthétase (HMGCS2)
- Hyperinsulinisme par déficit en 3-hydroxylacyl-CoA déshydrogénase des acides gras à chaîne courte (HADH)
- Déficit en acyl-CoA déshydrogénase des acides gras à chaîne moyenne (ACADM)
- Déficit en bêta-cétothiolase (ACAT1)
- Argininémie (ARG1)
- Déficit en décarboxylase des acides aminés aromatiques (DDC)
- Déficit en biotinidase (BTD)
- Déficit en carbamoyl-phosphate synthétase I (hyperammoniémie) (CPS1)
- Déficit systémique primaire en carnitine (SLC22A5)
- Déficit en carnitine palmitoyltransférase (CPT1A, CPT2)
- Céroïde-lipofuscinose neuronale (TPP1)
- Maladie de surcharge en esters de cholestérol (LIPA)
- Citrullinémie de type I (ASS1)
- Citrullinémie de type II (SLC25A13)
- Déficit primaire en coenzyme Q10 (COQ2)
- Trouble congénital de la glycosylation (MPI)
- Déficit en pyruvate déshydrogénase (DLD)
- Déficit en galactokinase / Galactosémie classique (GALK1, GALT)
- Syndrome de déficit en transporteur du glucose type 1 (SLC2A1)
- Acidurie glutarique de type I (GCDH)
- Glycogénose par déficit en glucose-6-phosphatase (AGL, G6PC, PHKG2, PYGL, SLC37A4)
- Atrophie gyrée choriorétinienne (OAT)
- Intolérance au fructose héréditaire / Déficit en fructose-1,6-diphosphatase (ALDOB, FBP1)
- Homocystinurie par déficit en cystathionine bêta-synthase (CBS, MTHFR, MTR, MTRR)
- Déficit en holocarboxylase synthétase (HLCS)
- Hypercalcémie infantile (CYP24A1, SLC34A1)
- Hypercholestérolémie familiale (APOB)
- Syndrome HHH (hyperornithinémie-hyperammoniémie-homocitrullinurie) (SLC25A15)
- Hyperoxalurie primitive (AGXT, GRHPR)
- Hyperphénylalaninémie (forme BH4-dépendante) (GCH1, PTS, QDPR)
- Hyperphénylalaninémie par déficit en tétrahydrobioptérine (DNAJC12)
- Acidémie méthylmalonique avec homocystinurie (ABCD4, LMBRD1, MMAA, MMAB, MMACHC, MMADHC, MMUT, PRDX1)
- Acidémie isovalérique (IVD)
- Maladie de Krabbe (GALC)
- Déficit congénital en lactase (LCT)
- Déficit en 3-hydroxyacyl-CoA déshydrogénase des acides gras à chaîne longue (HADHA)
- Intolérance aux protéines dibasiques avec lysinurie (SLC7A7)
- Maladie des urines sirop d'érable (BCKDHA, BCKDHB, DBT)
- Leucodystrophie métachromatique (ARSA)
- Mucopolysaccharidose (ARSB, IDS, IDUA)
- Déficit multiple en acyl-CoA déshydrogénases (ETFA, ETFB, ETFDH)
- Hyperammoniémie par déficit en N-acétylglutamate synthase (NAGS)
- Déficit en ornithine transcarbamylase (hyperammoniémie)(OTC)
- Phénylcétonurie (PAH)
- Glycogénose par déficit en phosphorylase kinase hépatique (PHGDH)
- Porphyrie érythropoïétique congénitale (FECH, UROS)
- Acidémie propionique (PCCA, PCCB)
- Troubles de la biosynthèse de la sérine (PSAT1)
- Déficit en succinyl-CoA:3-oxoacide CoA transférase (OXCT1)
- Déficit congénital en saccharase-isomaltase (SI)
- Polyneuropathie progressive avec nécrose striatale bilatérale (SLC25A19)
- Tyrosinémie (FAH, TAT)
Maladies des reins (néphrologie)
- Cystinose néphropathique (CTNS)
- Diabète insipide néphrogénique (AQP2, AVPR2)
- Acidose tubulaire distale (ATP6V0A4, ATP6V1B1)
- Syndrome néphrogénique d’antidiurèse inappropriée (AVPR2)
- Syndrome néphrotique corticorésistant héréditaire (COQ8B, NPHS1)
- Pseudo-hypoaldostéronisme (NR3C2)
- Pseudohypoparathyroïdisme (GNAS)
Maladies du cerveau et du système nerveux (neurologie)
- Ataxie par déficit en vitamine E (TTPA)
- Maladie des noyaux gris centraux sensible à la biotine et à la thiamine (SLC19A3)
- Syndrome de Brown-Vialetto-Van Laere / Déficit en transporteur de la riboflavine (SLC52A2, SLC52A3)
- Syndrome myasthénique congénital (AGRN, CHAT, CHRNA1, CHRNB1, CHRND, CHRNE, COL13A1, COLQ, DOK7, DPAGT1, GFPT1, MUSK, PREPL, RAPSN, SCN4A, SLC25A1, SLC5A7, SNAP25, SYT2)
- Encéphalopathie développementale et épileptique pyridoxino-dépendante (ALDH7A1, PLPBP)
- Paralysie périodique hypokaliémique de type 1 (CACNA1S)
- Déplétion de l'ADN mitochondrial, forme myopathique (TK2)
- Syndrome neurodégénératif dû au déficit de transport cérébral des folates (FOLR1)
- Maladie de Pompe (GAA)
- Encéphalopathie développementale et épileptique pyridoxal-phosphate dépendante (PNPO)
- Amyotrophie spinale infantile de type I (SMN1)
Maladies liées au cancer (oncologie)
- Rétinoblastome (RB1)
Maladies des poumons et de la respiration (pneumologie)
- Mucoviscidose (CFTR)
- Protéinose alvéolaire pulmonaire (forme Réunion) (MARS1)
Troubles sensoriels
- Syndrome branchio-otique (EYA1, SIX1)
- Surdité (ACTG1, CABP2, CDH23, CIB2, CLDN14, CLIC5, ESPN, ESRRB, GIPC3, GJB2, GJB6, GRXCR1, GRXCR2, HGF, ILDR1, KCNQ4, LHFPL5, LOXHD1, LRTOMT, MARVELD2, MSRB3, MYO15A, MYO6, MYO7A, NARS2, OTOA, OTOF, OTOG, OTOGL, PCDH15, PDZD7, PJVK, POU3F4, PRPS1, PTPRQ, RDX, RIPOR2, S1PR2, SIX1, SLC26A4, SLITRK6, STRC, SYNE4, TBC1D24, TECTA, TMC1, TMIE, TMPRSS3, TPRN, TRIOBP, USH1C, WFS1, WHRN)
- Surdité avec aplasie du labyrinthe, microtie et microdontie (FGF3)
- Surdité congénitale avec onychodystrophie (dominant autosomique) (ATP6V1B2)
- Glaucome congénital primaire (type 3) (CYP1B1, LTBP2)
- Amaurose congénitale de Leber (type 2) (RPE65)
- Maladie de Norrie (NDP)
- Syndrome de Pendred (SLC26A4)
- Rétinite pigmentaire (RPE65)
- Syndrome de Stickler (COL11A1, COL2A1)
- Syndrome d’Usher (ADGRV1, CDH23, CLRN1, MYO7A, PCDH15, USH1C, USH1G, USH2A, WHRN)
- Syndrome de Waardenburg (KITLG)
- Syndrome de Wolfram-like (dominant autosomique) (WFS1)
Maladies des os et du squelette (troubles squelettiques)
- Syndrome de brachydactylie-hypertension artérielle (PDE3A)
- Hypoparathyroïdie familiale isolée (GCM2, PTH)
- Hypophosphatasie de l’enfant (ALPL)
- Rachitisme hypophosphatémique (DMP1, FGF23, PHEX)
- Syndrome de Kenny-Caffey (type 2) (FAM111A)
- Chondrodysplasie métaphysaire de type Jansen (PTH1R)
- Ostéopétrose avec acidose tubulaire rénale (CA2, CLCN7, SNX10, TCIRG1, TNFSF11)
- Calcinoses tumorales familiales hyperphosphatémiques (FGF23, KL)
- Rachitisme hypocalcémique vitamine D-dépendant (CYP27B1, CYP2R1, CYP3A4, VDR)
Maladies du coeur et de la circulation sanguine (cardiovasculaire)
- Syndrome d'Andersen-Tawil (KCNJ2)
- Anomalie du septum atrial type ostium secundum (NKX2-5)
- Cardiomyopathie dilatée (MYH7)
- Cardiomyopathie hypertrophique familiale (MYH7)
- Syndrome d’Ehlers-Danlos vasculaire (COL3A1)
- Syndrome du QT long (CACNA1C)
Maladies de la peau (dermatologie)
- Syndrome de Griscelli de type 2 (RAB27A)
- Insensibilité congénitale à la douleur (récessif autosomique) (SCN9A)
- Syndrome de Rothmund-Thomson (RECQL4)
Troubles hormonaux (endocrinologie)
- Syndrome triple A (AAAS)
- Hyperplasie congénitale des surrénales par déficit en 17-alpha-hydroxylase (CYP11B1, CYP17A1)
- Obésité par déficit du récepteur de la mélanocortine 4 (MC4R)
- Complexe de Carney (PRKAR1A)
- Syndrome cerveau-poumon-thyroïde (NKX2-1)
- Hypoaldostéronisme familial (CYP11B2)
- Syndrome de Denys-Drash (WT1)
- Diabète néonatal permanent isolé (GCK)
- Syndrome de DiGeorge (TBX1)
- Hyperplasie congénitale des surrénales par déficit en cytochrome P450 oxydoréductase (POR)
- Syndrome de Wolcott-Rallison (EIF2AK3)
- Syndrome de Fanconi atypique-hyperinsulinisme néonatal (HNF4A)
- Déficit hypohysaires multiples de cause génétique identifiée (FOXA2)
- Déficit isolé familial en glucocorticoïdes (TXNRD2)
- Hyperinsulinisme par déficit en 3-hydroxylacyl-CoA déshydrogénase des acides gras à chaîne courte (HADH)
- Hyperlipoprotéinémie de type I et V(LPL)
- Hyperparathyroïdie isolée familiale (CDC73)
- Hypertriglycéridémie (APOA5)
- Déficit en apolipoprotéine A-I (ABCA1, APOA1)
- Hypobétalipoprotéinémie (APOB)
- Hypocalcémie autosomique dominante (GNA11)
- Syndrome IMAGe (CDKN1C)
- Insuffisance somatotrope isolée type IA et IV (GH1, GHRHR)
- Obésité par déficit congénital en leptine (LEP, LEPR)
- Lipodystrophie généralisée congénitale (AGPAT2, BSCL2, CAV1, CAVIN1)
- Syndrome MIRAGE (SAMD9)
- Obésité par déficit en pro-opiomélanocortine (POMC)
- Hypothyroïdie par déficit en facteurs de transcription impliqués dans le développement ou la fonction hypophysaire (LHX3, POU1F1, PROP1)
- Syndrome de Schaaf-Yang (MAGEL2)
- Histiocytose non langerhansienne (APOE)
- Syndrome SHORT (PIK3R1)
- Résistance aux hormones thyroïdiennes par mutation du récepteur aux hormones thyroïdiennes bêta (THRB)
- Syndrome de Wolfram type 1 (WFS1)
- Syndrome d’Antley-Bixler avec anomalies génitales et troubles de la stéroïdogenèse (POR)
- Syndrome hypoparathyroïdie, surdité neurosensorielle et dysplasie rénale (GATA3)
Maladies de l'estomac et des intestins (gastroentérologie)
- Syndrome d’Alagille de type 1 (JAG1)
- Déficit en alpha-1-antitrypsine (SERPINA1)
- Déficit en conjugaison des acides biliaires de type 1 (BAAT)
- Cholestase intrahépatique familiale progressive (NR1H4)
- Syndrome de grêle court congénital (CLMP)
- Diarrhée congénitale (NEUROG3)
- Syndrome de Dubin-Johnson (ABCC2)
- Syndrome d'hypopéristaltisme intestinal-microcôlon-mégavessie (ACTG2)
- Syndrome d'hypoplasie du pancréas-atrésie intestinale-hypoplasie de la vésicule biliaire (RFX6)
- Agénésie partielle pancréatique (PTF1A)
- Syndrome de Shwachman-Diamond (EFL1, SBDS)
Maladies du sang et du système immunitaire (hématologie / immunologie)
- Afibrinogénémie congénitale (FGA, FGB, FGG)
- Alpha-thalassémie (HBA1, HBA2)
- Analbuminémie congénitale (ALB)
- Anémie sidéroblastique autosomique récessive (SLC25A38)
- Angio-oedème héréditaire avec C1Inh normal lié à PLG (PLG)
- Thrombophilie héréditaire due au déficit congénital en antithrombine (SERPINC1)
- Calcification artérielle généralisée infantile (ABCC6, ENPP1)
- Atransferrinémie congénitale(TF)
- Syndrome lymphoprolifératif auto-immun (FAS, PRKCD)
- Polyendocrinopathie auto-immune type 1 (AIRE)
- Syndrome auto-inflammatoire-fièvre périodique-entérocolite infantile (NLRC4)
- Syndrome de Blau (NOD2)
- Déficit isolé de stockage des granules plaquettaires delta (FLI1, RASGRP2)
- Syndrome de Bloom (RECQL3)
- Syndrome de Coats-plus (STN1)
- Ostéomyélite stérile multifocale avec périostéite et pustulose (IL1RN)
- Syndrome CINCA (NLRP3)
- Déficit immunitaire par déficit des composés terminaux de la voie classique du complément (C8A)
- Anémie de Diamond-Blackfan (RPL35)
- Dyskératose congénital (ACD, DCLRE1B, DKC1, NHP2, NOP10, TINF2, WRAP53)
- Dysplasie ectodermique hypohydrotique avec déficit immunitaire (NFKBIA)
- Syndrome familial auto-inflammatoire au froid associé à NLRC4 / Urticaire familiale au froid (NLRC4, NLRP3)
- Fièvre méditerranéenne familiale (autosomique récessive) (MEFV)
- Anémie de Fanconi (BRCA1, BRCA2, BRIP1, ERCC4, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, MAD2L2, PALB2, RAD51, RAD51C, RFWD3, SLX4, UBE2T, XRCC2)
- Syndrome de dérèglement immunitaire-maladie inflammatoire de l'intestin-arthrite-infections récurrentes (TTC7A)
- Déficit en glutathion synthétase (GSS)
- Syndrome des plaquettes grises (NBEAL2)
- Hémoglobinose H (HBA1)
- Anémie hémolytique par déficit en glutathion réductase (GSR)
- Syndrome de fièvre périodique avec hyperimmunoglobulinémie D (MVK)
- Syndrome hyper-IgE avec infections récurrentes (STAT3, STAT6, ZNF341)
- Syndrome d’Imerslund-Gräsbeck (AMN, CUBN)
- Syndrome lymphoprolifératif auto-immun dû à une haploinsuffisance de CTLA-4 (CTLA4)
- Immunodéficience (AICDA, ARHGEF1, BCL10, CARMIL2, CD247, CD4, CDCA7, CTPS1, GATA2, IFIH1, IKBKG, LAT, LIG1, LRBA, RASGRP1, REL, RELB, STAT1, TFRC, UNG, ZNFX1)
- Maladie inflammatoire chronique de l’intestin (IL10RA, IL10RB)
- Déficit en facteur intrinsèque (CBLIF)
- Déficit d'adhésion leucocytaire (FERMT3)
- Syndrome lymphoprolifératif (CD27, CD70, ITK, XIAP)
- Thrombocytopénie syndromique liée à MYH9 (MYH9)
- Déficit du complexe majeur d’histocompatibilité (TAP1)
- Déficit en mévalonate kinase (MVK)
- Syndrome de Muckle-Wells (NLRP3)
- Neutropénie sévère congénitale (CSF3R, ELANE, GFI1, JAGN1, SRP54, VPS45, WAS)
- Déficit congénital en plasinogène (PLG)
- Poïkilodermie avec neutropénie (USB1)
- Déficit en properdine (CFP)
- Déficit congénital en prothrombine (F2)
- Syndrome pseudo-TORCH (STAT2, USP18)
- Syndrome de fibrose pulmonaire ± aplasie médullaire (lié aux télomères) (NAF1, PARN, POT1, RPA1, RTEL1, TERC, TERT, ZCCHC8)
- Syndrome d'arthrite purulente-pyoderma gangrenosum-acné / Syndrome PAPA (PSTPIP1)
- Syndrome plaquettaire du Québec (PLAU)
- Syndrome de synostose radio-ulnaire-thrombocytopénie amégacaryocytique (HOXA11)
- Déficit combiné facteur V et VIII (F11, F5, MCFD2)
- Syndrome de Roifman (RNU4ATAC)
- Syndrome de dystrophie rétinienne-oedème du nerf optique-splénomégalie-anhidrose-céphalée migraineuse (ALPK1)
- Dysplasie immuno-osseuse de Schimke (SMARCAL1)
- Déficit immunitaire/Immunodéficience combiné lié à RAC2 (RAC2)
- Anémie mégaloblastique thiamine-dépendante (SLC19A2)
- Thrombopénie (GNE, RAP1B, RBM8A)
- Thrombocytopénie liée à l'X avec plaquettes normales (WAS)
- Thrombophilie héréditaire sévère due au déficit congénital en protéine C / en protéine S (PROC, PROS1)
- Déficit en adénosine désaminase 2 (ADA2)
- Déficit combiné en facteurs de coagulation dépendants de la vitamine K de type 1 (GGCX)
- Maladie de von Willebrand (VWF)
- Syndrome WHIM (CXCR2, CXCR4)
Maladies du métabolisme
- Acrodermatite entéropathique (déficit en zinc, forme héréditaire)(SLC39A4)
- Déficit en acyl-CoA déshydrogénase des acides gras à chaîne très longue (ACADVL)
- Déficit en adénosine phosphoribosyltransférase (APRT)
- Alcaptonurie (HGD)
- Acidurie argininosuccinique (ASL)
- Syndrome de Barth (TAFAZZIN)
- Syndrome d'autisme-épilepsie par déficit en kinase déshydrogénase des cétoacides à chaînes ramifiées (BCKDK)
- Syndrome de déficit cérébral en créatine (GATM)
- Trouble de la biosynthèse du coenzyme Q10 (COQ6, COQ7)
- Syndromes myasthéniques congénitaux par défaut de glycosylation/ Anomalie congénitale de la glycosylation associée à PGM1/ Anomalie congénitale de la glycosylation type IIn (DPAGT1, PGM1, SLC39A8)
- Déficit en dihydropyrimidine déshydrogénase (DPYD)
- Fucosidose (FUCA1)
- Galactosémie type 3 (GALE)
- Malabsorption glucose-galactose (SLC5A1)
- Maladie de stockage du glycogène (GBE1, GYS1, GYS2, PFKM, PHKA2, PHKB)
- Encéphalopathie hyperammoniémique par déficit en anhydrase carbonique VA (CA5A)
- Encéphalopathie associée à une hyperméthioninémie par déficit en adénosine kinase (ADK)
- Hyperoxalurie primitive (HOGA1)
- Hypercalcémie hypocalciurique familiale V (AP2S1, CASR, GNA11)
- Alpha-mannosidose (MAN2B1)
- Déficit en acyl-CoA déshydrogénase 9 (ACAD9)
- Déficit en protéine trifonctionnelle mitochondriale (HADHA, HADHB)
- Mucolipidose (MCOLN1)
- Mucopolysaccharidose type 4A et type 7 (GALNS, GUSB)
- Maladie de Niemann-Pick (NPC1, NPC2)
- Déficit en pyruvate déshydrogénase E1-alpha (PDHA1)
- Encéphalopathie de l'enfant par déficit en thiamine pyrophosphokinase (TPK1)
- Maladie de Wilson (ATP7B)
- Maladie de Wolman (LIPA)
Maladies des reins (néphrologie)
- Syndrome d’Alport (COL4A3, COL4A4, COL4A5)
- Excès apparent en minéralocorticoïdes (HSD11B2)
- Syndrome de Bartter (BSND, CLCNKB, KCNJ1, SLC12A1)
- Cystinurie (SLC3A1, SLC7A9)
- Syndrome de Fanconi-Bickel (SLC2A2)
- Tubulopathie hypokaliémique avec surdité (KCNJ16)
- Hypomagnésémie (CLDN16, CLDN19, TRPM6)
- Pseudo-hypoaldostéronisme (SCNN1A, SCNN1B, SCNN1G)
- Pseudohypoparathyroïdisme (STX16)
- Néphropathie tubulo-interstitielle autosomique dominante associée à HNF1B (HNF1B)
- Acidose tubulaire rénale distale avec anémie (SLC4A1)
- Syndrome néphrotique familial corticorésistant avec insuffisance surrénalienne/SPLIS (SGPL1)
- Syndrome d'amélogenèse imparfaite-néphrocalcinose (FAM20A)
Maladies du cerveau et du système nerveux (neurologie)
- Syndrome d’Aicardi-Goutières (ADAR, IFIH1, LSM11, RNASEH2A, RNASEH2B, RNASEH2C, RNU7, SAMHD1, TREX1)
- Syndrome d’Allan-Herndon-Dudley (SLC16A2)
- Syndrome de déficit cérébral en créatine (GAMT)
- Xanthomatose cérébrotendineuse (CYP27A1)
- Maladie de Charcot-Marie-Tooth démyélinisante / CMT1A (PMP22)
- Dystonie sensible à la L-Dopa (SPR)
- Dystrophie musculaire d’Emery-Dreifuss (type 2, autosomique dominant) (LMNA)
- Hyperekplexie (GLRA1, GLRB, SLC6A5)
- Paralysie périodique hyperkaliémique (SCN4A)
- Hyperthermie maligne de l'anesthésie (CACNA1S, RYR1)
- Dystrophie musculaire de Duchenne (DMD)
- Syndrome muscle-oeil-cerveau (GMPPB)
- Myopathie distale de type 4 (FLNC)
- Filaminopathie musculaire (DES, FLNC)
- Myotonie fluctuante (SCN4A)
- Neuronopathie motrice distale héréditaire (COQ7, SORD)
- Paramyotonie congénitale (SCN4A)
- Syndrome de Prader-Willi (SNRPN)
- Syndrome d'hyperaldostéronisme primitif-épilepsie-anomalies neurologiques (CACNA1D)
- Syndrome de Schwartz-Jampel de type 1 (HSPG2)
- Syndrome de convulsions infantiles-choréoathétose (PRRT2)
- Sclérose tubéreuse de Bourneville (TSC1, TSC2)
- Myopathie viscérale familiale (ACTG2)
Maladies liées au cancer (oncologie)
- Polypose adénomateuse familiale du côlon (APC)
- Polypose juvénile associée à la télangiectasie hémorragique héréditaire (SMAD4)
- Syndrome de Li-Fraumeni (TP53)
- Syndrome de réparation des mésappariements (mismatch repair)(MLH1, MSH2, MSH6, PMS2)
- Syndrome de Nijmegen (NBN)
- Syndrome de Peutz-Jeghers (STK11)
- Blastome pleuropulmonaire (DICER1)
- Syndrome de von Hippel-Lindau (VHL)
- Néphroblastome (WT1)
Maladies des poumons et de la respiration (pneumologie)
- Dysplasie alvéolo-capillaire congénitale (FOXF1)
- Syndrome d'Ondine (PHOX2B)
- Dyskinésie ciliaire primitive (CCDC103, CCDC39, CCDC40, CCDC65, CCNO, CFAP298, CFAP300, DNAAF1, DNAAF2, DNAAF3, DNAAF4, DNAAF5, DNAH11, DNAH5, DNAH9, DNAI1, DNAI2, DNAJB13, DRC1, FOXJ1, GAS2L2, GAS8, HYDIN, LRRC6, MCIDAS, NEK10, NME5, ODAD1, ODAD2, ODAD3, ODAD4, PIH1D3, RSPH1, RSPH3, RSPH4A, RSPH9, SPAG1, TP73, TTC12, ZMYND10)
- Syndrome coxo-podo-patellaire (TBX4)
- Syndrome de dyskinésie ciliaire primitive-rétinite pigmentaire (RPGR)
- Trouble du métabolisme du surfactant pulmonaire (ABCA3, CSF2RA, SFTPB, SFTPC)
Troubles sensoriels
- Syndrome d’Alström (ALMS1)
- Syndrome de Bardet-Biedl (BBS1, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, CEP290, MKKS)
- Syndrome avec ataxie cérébelleuse, aréflexie, pied creux, atrophie optique et surdité neurosensorielle (ATP1A3)
- Syndrome de Chudley-McCullough (GPSM2)
- Surdité rare neurosensorielle non syndromique autosomique récessive type DFNB/ Surdité génétique non syndromique (PNPT1)
- Syndrome d'hypoplasie médiofaciale-surdité-elliptocytose-néphrocalcinose (AMMECR1)
- Syndrome d’ostéoporose-pseudogliome (LRP5)
- Syndrome de Stickler (COL9A1)
- Syndrome de Waardenburg (EDN3, EDNRB, MITF, PAX3, SOX10)
- Syndrome de la cornée fragile de type 1 (ZNF469)
Maladies des os et du squelette (troubles squelettiques)
- Achondroplasie (FGFR3)
- Acrodysostose de type 2 avec ou sans résistance hormonale (PDE4D)
- Syndrome de Bruck de type 2 (PLOD2)
- Hypoplasie cartilage-cheveux (RMRP)
- Syndrome d’Ehlers-Danlos (ADAMTS2, AEBP1, B3GALT6, B4GALT7, C1R, C1S, COL1A1, COL1A2, COL5A1, COL5A2, DSE, FKBP14, PLOD1, SLC39A13, TNXB)
- Fibrodysplasie ossifiante progressive (ACVR1)
- Rachitisme hypophosphatémique héréditaire avec hypercalciurie (SLC34A3)
- Ostéogenèse imparfaite (BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, SERPINF1)
- Ostéopétrose (OSTM1, TNFRSF11A)
- Dysplasie osseuse ostéosclérotique (FAM20C)
- Syndrome de calcinose tumorale hyperphosphatémique familiale (GALNT3)